Computergestützte Modellierung biologischer Prozesse
Forschungsbericht (importiert) 2009 - Max-Planck-Institut für Biophysik
Einführung
Die Erstellung mathematischer Modelle ist ein dreistufiges Verfahren. Im ersten Schritt müssen die Randbedingungen des Modells definiert werden. Dazu gehören die Bestimmung der Modellkomponenten; bei biologischen Modellen sind das in der Regel Proteine, Metabolite, Gene und deren Initialwerte. Im zweiten Schritt werden die Interaktionen zwischen den Modellkomponenten festgelegt, also das Reaktionsnetzwerk des Modells. Im dritten Schritt erfolgt die Belegung der Modellreaktionen mit kinetischer Information, das heißt mit den mathematischen Gesetzen und kinetischen Parametern, die die Dynamik des Modells bestimmen [1].
Die Arbeitsgruppe Bioinformatik der Abteilung Analyse des Vertebratengenoms am Max-Planck-Institut für molekulare Genetik beschäftigt sich bereits seit einigen Jahren mit der Entwicklung von Software und Methodologie zur Erstellung derartiger mathematischer Modelle. Diese dienen einer Vielzahl von Anwendungen, zum Beispiel der Entwicklung von Krankheitsmodellen für verschiedene Tumortypen, für Typ-2 Diabetes mellitus, für Modelle zur Beschreibung der Zellantwort auf Chemikalien sowie für Modelle zur Entwicklungsbiologie im Allgemeinen.
Markeridentifizierung
Krankheiten können durch so genannte molekulare Marker (Gene, Proteine, Metabolite) im Labor diagnostiziert werden. Die Identifizierung spezifischer molekularer Marker, zum Beispiel zur frühzeitigen Diagnose des Krankheitsausbruchs, ist ein wichtiger Bestandteil der Genomforschung. Zur Markeridentifizierung werden zumeist statistische Verfahren der Bioinformatik eingesetzt. Ein typischer Ansatz besteht im statistischen Vergleich (Hypothesentest) von verschiedenen Zuständen, beispielsweise „erkrankt“ gegen „gesund“ oder „behandelt“ gegen „unbehandelt“ [2]. Wenn wenig Vorwissen vorhanden ist, werden dazu im Allgemeinen genomweite experimentelle Techniken wie microarrays oder Hochdurchsatz-Sequenzierungen bei der Genexpressionsanalyse genutzt. Bioinformatische Verfahren zur Markeridentifizierung sind dabei eng mit der Prozessierung der Daten (Qualitätskontrolle, Normalisierung) verbunden. Statistische Testverfahren werden benutzt, um die Signifikanz der Änderungen der Markerexpression beim Vergleich der verschiedenen Zustände zu bewerten.
Neben diesen Ansätzen für Einzelvergleiche kommen immer mehr auch integrative Ansätze zum Einsatz. Diese Ansätze beziehen die zahlreichen öffentlich verfügbaren Datensätze in die Analyse mit ein (Abb. 1). Die in Einzelvergleichen gewonnene Information kann so auf ihre Allgemeingültigkeit hin überprüft werden, um möglichst robuste Marker zu identifizieren [3].
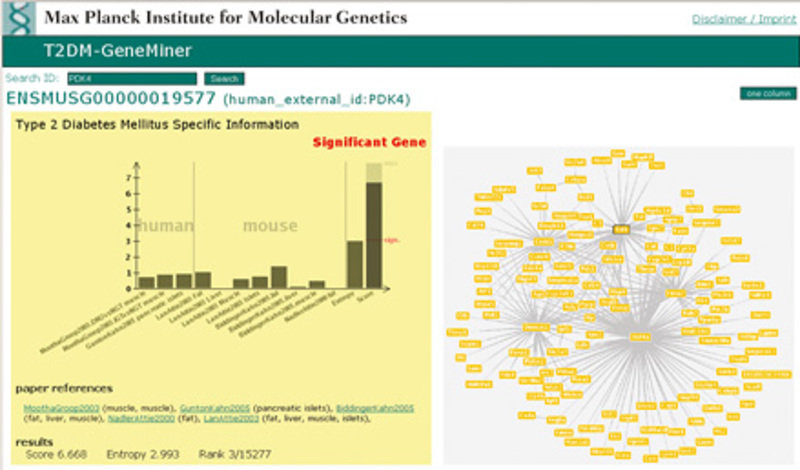 Bild vergrößern
Bild vergrößern
Integration humaner funktioneller Netzwerke
Es existiert bereits eine Vielzahl (> 200) öffentlich zugänglicher Datenbanken, in denen biologische Prozesse annotiert und visualisiert werden. Diese Datenbanken (beispielsweise KEGG [4] oder Reactome [5]) bilden die Basis zur Bestimmung der Netzwerke, die die Modellkomponenten miteinander verbinden. Ein fundamentaler Nachteil in der Bestimmung der Reaktionsnetzwerke besteht in der Diversität der Datenbankannotationen, also der Tatsache, dass Datenbanken oft nur einen speziellen Datentyp abdecken, zum Beispiel Daten für Protein-Protein- Interaktionen, für metabolische Reaktionen oder für Signalwege. Biologische Prozesse umfassen jedoch üblicherweise mehrere dieser Datentypen. Dies führt in der Praxis dazu, dass ein Anwender viele verschiedene Datenbanken abrufen muss, um die nötige Information über das Reaktionsnetzwerk zu bekommen. In der Arbeitsgruppe Bioinformatik wurde deshalb die integrative Datenbank ConsensusPathDB entwickelt, die die verschiedenen Interaktionstypen integriert und eine umfassende Annotation ermöglicht [6]. ConsensusPathDB bündelt zurzeit den Inhalt von 12 primären Datenbanken für humane funktionelle Interaktionen. Sie enthält Information über 25831 verschiedene Komponenten (Proteine, Gene, Metabolite etc.) und 73426 verschiedene funktionelle Interaktionen und deckt dabei 1689 humane biologische Prozesse ab (zum Beispiel den Insulin-Signalweg, Glykolyse etc.). Diese Integration wurde ermöglicht durch die Entwicklung eines einheitlichen Datenbankschemas, das in der Lage ist, die verschiedenen Typen von Interaktionen und Modellkomponenten zu integrieren. ConsensusPathDB ist im Internet über einen Webserver frei zugänglich. Verschiedene Funktionalitäten erlauben beispielsweise die Suche nach Reaktionen von ausgewählten Proteinen, nach kürzesten Wegen zwischen zwei Proteinen, nach Methoden zur Identifizierung angereicherter Prozesse und nach Visualisierungskomponenten. Netzwerke können in den gängigen Formaten PSI-MI, BioPAX und SBML importiert und exportiert werden, was eine weitgehende Kompatibilität mit anderen Systemen ermöglicht (Abb. 2).
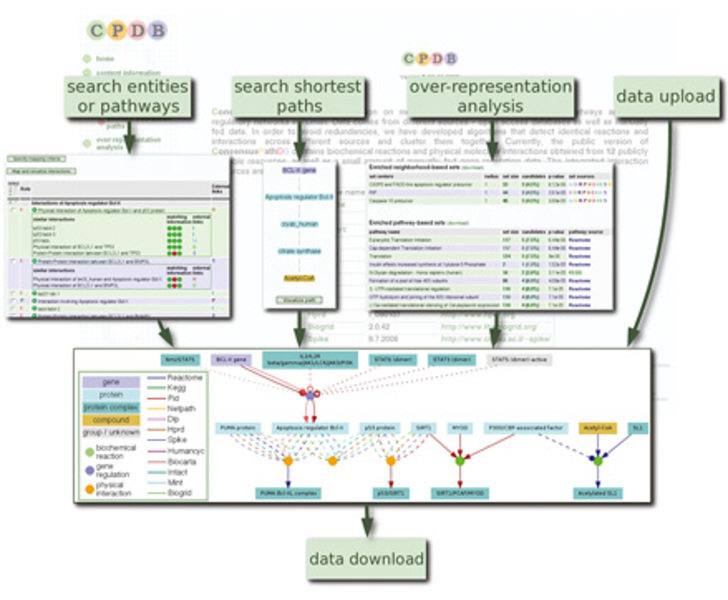 Bild vergrößern
Bild vergrößern
Modellierung biologischer Prozesse mit PyBioS
Aufbauend auf der Struktur des Reaktions- und Interaktionsnetzwerks kann ein mathematisches Modell erstellt werden, welches beispielsweise durch ein gewöhnliches Differenzialgleichungssystem gegeben ist. Ein derartiges Modell beschreibt die zeitlichen Konzentrationsänderungen der Modellkomponenten. Für die Erstellung eines solchen Modells benötigt man zudem kinetische Gesetze, die die Geschwindigkeiten der einzelnen Reaktionen beschreiben. Beispiele für eine derartige Kinetik sind unter anderem die von Michaelis und Menten abgeleitete Kinetik für enzymkatalysierte Reaktionen oder die Massenwirkungskinetik. Die genaue Geschwindigkeit einer einzelnen Reaktion wird dabei durch kinetische Parameter und die Konzentrationen der beteiligten Reaktionspartner festgelegt. Kinetische Parameter müssen experimentell ermittelt oder an gemessene Zustandsänderungen angepasst werden. Vielfach ist es aber auch möglich, Näherungen für die kinetischen Modellparameter zu verwenden, um qualitative Aussagen über das Modellverhalten zu treffen.
Für die Erstellung mathematischer Modelle ist in der Arbeitsgruppe Bioinformatik das Computerprogramm PyBioS entwickelt worden [1,7,8]. PyBioS hat eine web-basierte Benutzerschnittstelle (Abb. 3). Das Programm automatisiert die Generierung und Simulation mathematischer Modelle und ermöglicht dadurch auch die Erstellung und Untersuchung großer Modelle. PyBioS automatisiert auch die Integration von Reaktionsnetzwerken aus Datenbanken wie KEGG, Reactome oder ConsensusPathDB und bietet eine Vielzahl vordefinierter kinetischer Gesetze an. Dies ermöglicht eine schnelle Modellerstellung und vermeidet Fehler in der Modellentwicklung. Ferner hat PyBioS auch vielfältige Funktionalitäten für die Visualisierung der Simulationsergebnisse, wie zum Beispiel die Darstellung von zeitlich verlaufenden Konzentrationsänderungen. Neben der reinen Simulation ist auch die Modellanalyse von großer Bedeutung. Hierfür bietet PyBioS beispielsweise die Möglichkeit, den Einfluss eines Modellparameters auf das Modellverhalten im stationären Zustand zu untersuchen. Eine derartige Sensitivitätsanalyse dient der Identifizierung kritischer Modellparameter.
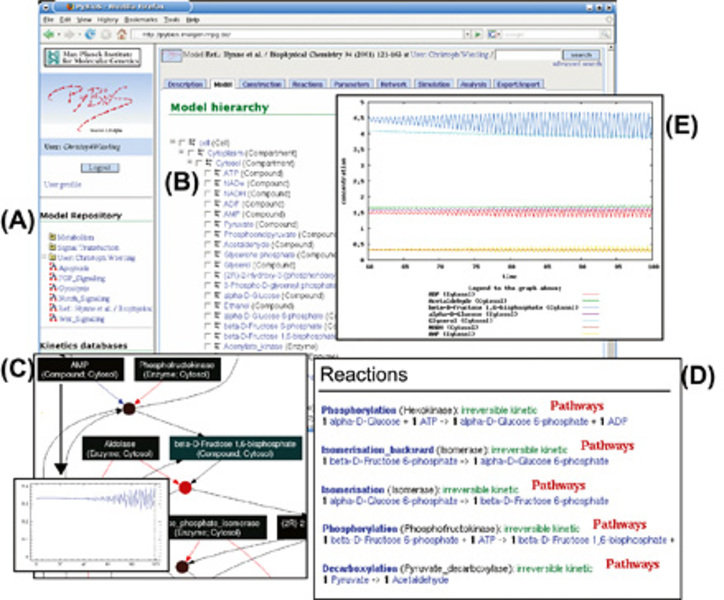 Bild vergrößern
Bild vergrößern
Zusammenfassung und Ausblick
In diesem Bericht wurden Werkzeuge für die verschiedenen Schritte bei der Erstellung und Analyse von computergestützten Modellen biologischer Prozessen vorgestellt. Diese Modelle ermöglichen eine Vorhersage des Verhaltens eines biologischen Systems,wenn zum Beispiel bestimmte Änderungen auftreten (Medikamentengabe, Umwelteinflüsse, Entwicklungsvorgänge). Die Modellierung von biologischen Prozessen, insbesondere von Krankheitsprozessen, besitzt ein hohes wissenschaftliches und wirtschaftliches Potenzial, ist jedoch zur Zeit noch zu generisch, um ein individuelles biologisches System, etwa das eines Patienten, exakt beschreiben zu können. Es wird in Zukunft darauf ankommen, gezielt molekulare Informationen in die mathematischen Modelle einzubauen, um beispielsweise Effekte von Mutationen oder individuelle Gen- und Proteinexpressionsdaten bei der Modellierung berücksichtigen zu können.