Interaktionsnetzwerke in Proteinstrukturen
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für Biophysik
Einführung
Neue Sequenzierungstechniken ermöglichen die Entschlüsselung der vollständigen genomischen Sequenz von immer mehr Organismen. Während vor zehn Jahren erst einige wenige Einzeller oder Viren sequenziert waren, sind inzwischen die DNA-Sequenzen hunderter verschiedener Arten vollständig verfügbar. Mittels experimenteller und bioinformatischer Methoden ist es möglich, Teilsequenzen zu bestimmen, die einzelne Proteine kodieren, und die genomische Sequenz direkt in eine Kette aus Aminosäuren zu übersetzen. Die Proteine unterscheiden sich in der Anzahl und Reihenfolge ihrer jeweiligen Aminosäuren, die verschiedene Seitengruppen tragen. Bislang ist es nicht möglich, im Voraus zu berechnen, wie sich die „eindimensionale“, linear gedachte Aminosäurekette in eine funktionierende Struktur faltet.
Das Faltungsproblem
Die Frage, wie sich die räumliche Struktur eines Proteins berechnen lässt, wird als das „Faltungsproblem“ bezeichnet. In der Zelle falten sich verschiedene Proteine je nach ihrer Größe und Sequenz unterschiedlich schnell - aber meist innerhalb eines Zeitraums von wenigen Sekunden bis hin zu einigen Minuten. Eine gefaltete Struktur befindet sich in einem energetischen Minimum. Leider hilft dieses Wissen aber nicht bei der Strukturvorhersage, was durch das Levinthal-Paradox verdeutlicht wird: Hierbei wird angenommen, man könne für ein Protein sämtliche physikalisch möglichen Konformationen simulieren und die Energie jeder Konformation innerhalb einer Millisekunde genau berechnen. Aufgrund der hohen Zahl an möglichen Konformationen würde es bereits für ein durchschnittlich kleines Protein (rund 100 Aminosäuren umfassend) mit dieser Suchmethode mehr Zeit in Anspruch nehmen, das Energieminimum zu finden, als das Universum alt ist.
Selbst für modernste Supercomputer sind aus diesem Grund quantenmechanische Berechnungen viel zu aufwändig, um vollständige Proteine, von physikalischen Grundprinzipien ausgehend, ab-initio zu berechnen. Um dennoch Strukturvorhersagen im Computer durchführen zu können, nimmt man die Tatsache zur Hilfe, dass sich ähnliche Aminosäuresequenzen zu ähnlichen Strukturen falten. Die so genannte Homologie-Modellierung überträgt die Informationen von experimentell aufgeklärten Strukturen auf neue Sequenzen. Die Methode versagt aber entsprechend, wenn keinerlei Proteinstruktur mit ausreichender Sequenzähnlichkeit bekannt ist. In der Regel muss eine bekannte Struktur mindestens 30 % Sequenzidentität aufweisen, um brauchbare Modelle zu liefern. Damit lassen sich aber nur für weniger als ein Drittel aller theoretisch aus der Gensequenz vorhergesagten menschlichen Proteine vollständige Modelle berechnen, für weniger als die Hälfte lässt sich immerhin zumindest ein Teil der Sequenz modellieren.
Die Lösung für das „schwere“ Ziel, ohne Sequenzähnlichkeit zu bekannten Strukturen eine neue Struktur zu bestimmen, scheint in so genannten „statistischen Potenzialen“ zu liegen. Hierbei werden Statistiken über die Kontakte zwischen zwei Aminosäuren gesammelt und mit den Werten verglichen, die man aufgrund der Häufigkeit dieser beiden Aminosäuren erwarten würde. Auf diese Weise lässt sich eine energetische Größe ableiten, aus der hervorgeht, wie „gerne“ zwei Aminosäuren miteinander in Wechselwirkung treten. Solche paarweise statistischen Potenziale haben zwar den Vorteil, dass sie auch auf Proteinsequenzen realistischer Größe (zwischen 100 und 200 Aminosäuren) angewandt werden können, jedoch die komplexen Zusammenhänge höherer Ordnung bei der Proteinfaltung (die Kooperativität) können sie nicht erklären. Daher ist es bislang nicht möglich, Proteinstrukturen de-novo vorherzusagen. Die erfolgreichsten Strukturvorhersageverfahren beruhen daher auf einer Kombination verschiedener sequenzbasierter Methoden und setzen statistische Potenziale nur zu einer abschließenden Feinabstimmung der resultierenden Modelle ein.
Ein neuer Ansatz
Der Umstand, dass Proteine, verglichen mit dem Levinthal-Paradoxon, außerordentlich schnell zu ihrer nativen räumlichen Struktur finden, bedeutet, dass sich Proteine zielgerichtet falten. Wissenschaftler gehen dabei von der Vorstellung aus, dass die Proteine sich ihrem Energieminimum nähern, ähnlich wie ein Ball in einer Schüssel zum tiefsten Punkt rollt. Bisherige Suchmethoden bleiben allerdings häufig in lokalen Minima stecken, da die energetische „Landschaft“ nicht glatt und monoton verläuft, sondern eher zerklüftet zu sein scheint. Die Wissenschaftler der Nachwuchsgruppe Bioinformatik / Strukturelle Proteomforschung entwickelten daher einen neuen Ansatz, bei dem die strukturelle Flexibilität der Proteine mit einbezogen und sogar aktiv genutzt wird. Faltungs- und Dockingproblem werden also nicht gesondert, sondern als eng verwandte Probleme, basierend auf gleichen molekularen Mechanismen, betrachtet.
Zentraler Ausgangspunkt der Arbeit ist, Proteinstrukturen als Netzwerk von Aminosäurekontakten darzustellen. Dabei werden die Strukturen in Graphen (Netzwerke) überführt, indem die einzelnen Aminosäuren („Residuen“) als Knoten und die Interaktionen zwischen ihnen als Kanten dargestellt werden. Die entstehenden Netzwerke werden als Residue-Interaction Graphs (RIGs) bezeichnet. RIGs haben entscheidende konzeptionelle Vorteile gegenüber einer geometrischen Betrachtung im dreidimensionalen Raum: Die gesamte Topologie des Netzwerkes kann in die Berechnungen mit einfließen, was bei einer paarweisen Betrachtung von Kontakten nicht möglich ist. Außerdem konnten bereits eine Reihe biophysikalischer Parameter mit Grapheigenschaften in Beziehung gesetzt werden [1]. Zudem haben Graphen den Vorteil, dass der Zustandsraum und somit Konformationsänderungen einer algorithmischen und graphentheoretischen Analyse wesentlich zugänglicher sind. Eine gängige und relativ einfach zu berechnende Definition von „Kontakt“ (Interaktion) basiert auf der Anwendung eines Distanz-Schwellwertes des Abstands zwischen Aminosäuren. Abbildung 1 zeigt einen Ausschnitt aus einer Proteinstruktur, bei der die zentralen Kohlenstoffatome (Cα) weniger als 8 Å voneinander entfernt liegen. Dadurch wird eine kugelförmige „Nachbarschaft“ direkter Kontakte um eine Aminosäure herum definiert. Besonders wichtig für die Bildung einer Struktur sind jedoch auch diejenigen Kontakte von Aminosäuren, die in der linear gedachten Sequenz weit auseinander liegen (Langstreckenkontakte). Abbildung 2 zeigt solche Langstreckenkontakte innerhalb eines Proteinkomplexes.
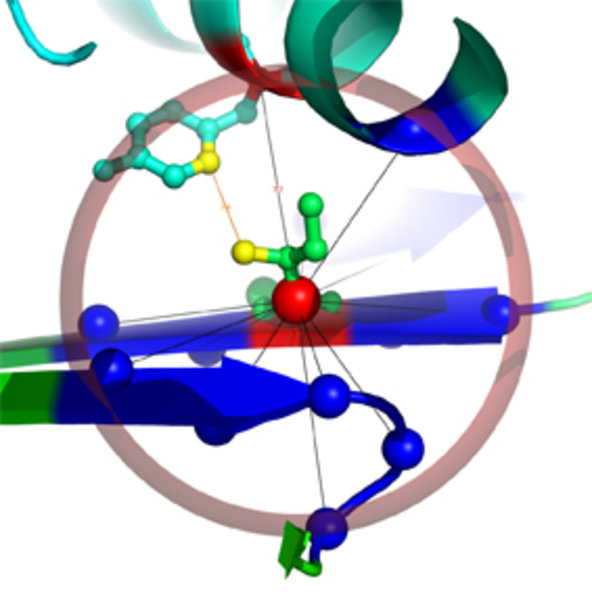 Bild vergrößern
Bild vergrößern
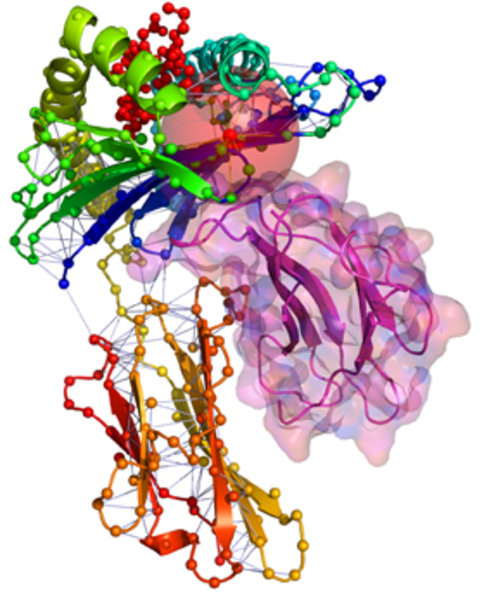 Bild vergrößern
Bild vergrößern
Natürlich sind Architektur und Eigenschaften der resultierenden Netzwerke zu einem gewissen Maß von der verwendeten Definition von „Kontakt” abhängig. Leider gibt es dafür bislang keine allgemeingültige Definition. In den bislang publizierten Studien variiert nicht nur der Schwellenwert, sondern auch, welche Atome in die Kontaktdefinition eingehen. Der Einfluss auf die resultierenden Netzwerke wird von den Wissenschaftlern systematisch untersucht. Die Datengrundlage ihrer Untersuchungen bildet die „ProteinDatenBank“ (PDB), in der alle bis dato veröffentlichten Proteinstrukturen gespeichert und öffentlich zugänglich sind - inzwischen über 40000 Strukturen mit über 10 Millionen Aminosäuren. Die meisten Kontaktdefinitionen ergeben im Schnitt 7-8 Kontakte pro Aminosäure, also insgesamt über 70 Millionen „Kanten“.
Analyse von Residue-Interaction Graphs (RIGs)
Die durch Übertragung einer Proteinstruktur in RIGs gewonnene Netzwerkinformation lässt sich vielfältig analysieren. Die Wissenschafter haben dafür innovative Software zur Netzwerk-Analyse von Proteinstrukturen, Interfaces und Konformationsänderungen entwickelt. An ihren Ergebnissen fällt auf, dass sich innerhalb eines Proteins Teilnetze unterscheiden lassen, die ganz unterschiedliche Eigenschaften aufweisen. Während die kompletten RIGs einem klassischen Zufallsgraphen ähnlich sehen, ist das Netzwerk der Langstreckenkontakte in seinem Aufbau Protein-Protein-Interaktionsnetzwerken sehr ähnlich. Auch hier haben die meisten Residuen nur wenige Kontakte, während einige wenige hochgradig verbundene Aminosäuren das Netzwerk zusammenhalten (Abb. 3). Lappe und Kollegen ist es gelungen, zentrale Bereiche dieser Netzwerke zu identifizieren, die mit so genannten „Faltungs-Elementen“ korrelieren. So ist es ihnen möglich, split-sites vorherzusagen, also Teile der Sequenz, bei denen ein Schnitt immer noch zu einem faltbaren Protein führt [3].
 Bild vergrößern
Bild vergrößern
Der Vergleich von Proteinstrukturen kann als optimierte Überlappung zwischen RIGs beschrieben werden. In seiner theoretischen Form ist dies zwar ein nicht berechenbares Problem, aber mittlerweile sind hierzu schnelle Approximationsverfahren entwickelt worden [4]. Die Wissenschaftler können damit innerhalb von RIGs aktive Zentren, Bindestellen und allosterische Kommunikationspfade als Teilgraphen beziehungsweise Signalwege darstellen und haben Verfahren entwickelt, diese vorherzusagen. Die zentrale Frage der Vorhersage von Strukturen über RIGs läuft auf die Entwicklung statistischer Vielkörper-Potenziale hinaus, da paarweise Potenziale dieses hochdimensionale Problem bislang nicht lösen konnten.
Strukturvorhersagen
Die Nachwuchsgruppe beschäftigt sich weiterhin mit dem Problem, Proteinstrukturen über Kontaktkarten vorherzusagen. Zunächst wurde das Konzept der Homologie-Modellierung als Übertragung von Netzwerkinformation zwischen Sequenzen erfolgreich umgesetzt. Außerdem setzen die Wissenschaftler Verfahren ein, um aus dieser Netzwerk-Information mit hoher Genauigkeit die dreidimensionale Struktur zu rekonstruieren[5]. Strukturvorhersagen können also über Kontaktkartenvorhersage erfolgen. Dies führt zu einem relativ simplen, aber aufschlussreichen Experiment: Das aus einer experimentellen Struktur gewonnene Netzwerk wird zunehmend durch zufälliges Einfügen oder Löschen von Kontakten „verrauscht“ und der Einfluss dieser Änderungen auf die Qualität der rekonstruierten 3D-Struktur gemessen [6]. Dabei wird deutlich, dass der größte Teil der Netzwerkinformation redundant ist: Über 50 % aller Kontakte können gelöscht werden, ohne dass eine nennenswerte Abweichung von der nativen Konformation auftritt. Es reichen also bereits wenige richtige Kontakte aus, um die Struktur eines Proteins zu bestimmen. Umgekehrt erhält man bereits bei 3 % falscher Kontakte eine schlechtere Rekonstruktionsqualität als bei 90 % fehlender Kontaktinformation (Abb. 4). Es ist also wesentlich wichtiger, wenige Kontakte richtig vorherzusagen, als viele Kontakte mit geringer Genauigkeit zu bestimmen.
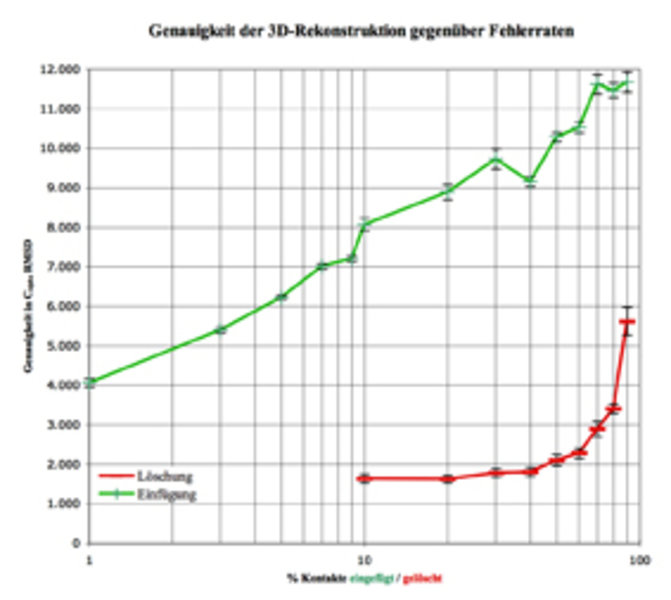 Bild vergrößern
Bild vergrößern
Durch die Weiterentwicklung dieses Verfahrens wollen die Wissenschaftler ermitteln, welche Knoten und Kontakte in solchen Strukturnetzwerken entscheidend sind. Dabei verfolgen sie interdisziplinäre Ansätze, bei denen Erkenntnisse aus mathematischen Gebieten wie Informationstheorie, Graphentheorie und Geometrie mit biophysikalischen und biochemischen Überlegungen kombiniert werden. Die gewonnen Erkenntnisse auf dem Gebiet der Proteinfaltung sollen die Grundlage für die Durchführung eines flexiblen Dockings bilden. Die Ergebnisse sind von Bedeutung für Fragen des Proteindesigns und der Entwicklung neuer Wirkstoffe.
Man ist zuversichtlich, mit der „Netzwerksicht“ auch weiterhin zur Lösung des Faltungs- und Dockingproblems sowie zur Funktionsvorhersage von Proteinen beitragen zu können [7, 8, 9]. Aufgrund ähnlicher Architekturen könnten auf der Basis der Strukturnetzwerke innerhalb der Protein-Protein-Interaktionsnetzwerke neue Vorhersage- und Analysemethoden entstehen, die den Herausforderungen neuer proteomischer Daten in Zukunft gerecht werden [10]. Aber die Wissenschaftler wagen auch einen vorsichtigen Blick über den Rand der Zelle hinaus - schließlich gibt es auch noch in anderen Bereichen Netzwerke (wie zum Beispiel das Internet, Netzwerke von Neuronen bis hin zu sozialen Netzwerken) mit ganz ähnlichen Architekturen und Eigenschaften, wo ihre Ergebnisse erfolgreich zur Anwendung kommen könnten.