A new approach for higher resolution structure determination of macromolecules in their native context
An optimized resolution in the subnanometer range by hybrid subtomogram averaging - single particle cryo-electron microscopy
Cryo-electron tomography that was combined with subtomogram averaging (StA) has yielded high-resolution structures of macromolecules in their native context. However, high-resolution StA is not commonplace due to beam-induced sample drift, images with poor signal-to-noise ratios (SNR), challenges in contrast transfer function (CTF) correction, and limited particle number. Here we address these issues by collecting tilt series with a higher electron dose at the zero-degree tilt. Particles of interest are then located within reconstructed tomograms, processed by conventional StA, and then re-extracted from the high-dose images in 2D. Single particle analysis tools are then applied to refine the 2D particle alignment and generate a reconstruction. Use of our hybrid StA (hStA) workflow improved the resolution for tobacco mosaic virus from 7.2 to 4.4 Å and for the ion channel Ryanodine receptor 1, RyR1 in crowded native membranes from 12.9 to 9.1 Å. These resolution gains make hStA a promising approach for other StA projects aimed at achieving subnanometer resolution.

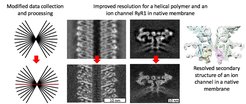
How the resolution is improved
Electron tomography of biological samples preserved in their hydrated form allows observing molecules in their native states such as intracellular proteins, membrane proteins or desoxyribonucleinacid (DNA). Molecules present in multiple copies in the tomographic volumes could be mutually aligned and averaged in a process called “subtomogram averaging”, allowing analysis of their structure and understanding the mechanisms of their functions. Yet, multiple challenges have to be dealt with in order to produce informative high-resolution structures: optical and geometrical distortions, laborious image processing and limited throughput during data collection.
On the other hand, a well-established method called "single-particle electron cryo-microscopy analysis (cryo-EM)" captures purified proteins frozen in a layer of amorphous ice in 2D and performs three-dimensional (3D) reconstruction from multiple protein copies viewed from different directions. Cryo-EM received wide recognition and the developers were acknowledged by a Nobel Prize in Chemistry in 2017. Single particle cryo-EM results in high-resolution structures but operates on macromolecules extracted from their native context which may lead to their non-native conformations.
Researchers from the Max Planck Institute of Biophysics in Frankfurt am Main suggested an improved workflow for structural determination from cryo electron tomograms by combining the advantages of subtomogram averaging and single particle cryo-EM. A single particle-like 2D image of an object of interest is followed by recording a tomogram. The macromolecules of interest are located in 3D tomograms and mutually aligned, however, the final refinement steps are performed using the single particle software from the single particle-like image. This workflow allows compensation for drift occurring during exposure of amorphous ice by electrons, and performing better reconstructions which eventually leads to higher resolution. This hybrid workflow also speeds up data collection allowing higher throughput on very expensive modern day cryo electron microscopes.
“We aimed at obtaining subnanometer reconstructions as at this resolution we can resolve alpha-helices of medically relevant membrane proteins with ion channels like sodium, potassium, or calcium channels. We could achieve improved resolution by modifying our data collection and processing routines and now we are happy to share our experience to the wider structural biology community” says research group leader Misha Kudryashev. “In the future we believe that this method will be used by us and the colleagues to gain unique insights into structure and function of macromolecules in their native context."